
Key Takeaways
- Ibrutinib is rapidly absorbed with a bioavailability of ~97% and reaches peak plasma levels in 1-2 hours.
- CYP3A4 is the primary enzyme responsible for its metabolism; strong inhibitors raise exposure dramatically.
- The drug’s effective half‑life is about 4-6 hours, but steady‑state is achieved after 3-4 days due to irreversible BTK binding.
- Food has minimal impact, but concomitant P‑gp substrates can alter intestinal absorption.
- Dosing adjustments are mainly needed for drug interactions and severe hepatic impairment.
When clinicians prescribe Ibrutinib is a covalent Bruton’s tyrosine kinase (BTK) inhibitor that has reshaped therapy for chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL). Understanding how the body handles the molecule-its pharmacokinetics (PK)-is essential for maximizing efficacy while limiting toxicity. This guide walks through absorption, distribution, metabolism, elimination, and the practical dosing nuances that arise from each step.
What Pharmacokinetics Means for a Drug
Pharmacokinetics is the study of how a drug moves through the body, encompassing four core processes: absorption, distribution, metabolism, and excretion (ADME). Each parameter influences the concentration‑time curve, which clinicians use to decide dose, frequency, and monitoring strategy.
Absorption: Getting Ibrutinib Into the Bloodstream
Ibrutinib is taken orally as a 140‑mg capsule or 420‑mg tablet. Clinical studies in healthy volunteers and patients with CLL report an absolute oral bioavailability of roughly 97%, meaning almost the entire dose reaches systemic circulation.
- Time to peak (Tmax): 1-2 hours after ingestion.
- Effect of food: High‑fat meals produce a modest 10% increase in AUC (area under the curve) but no clinically relevant change in efficacy; patients can therefore take the drug with or without food.
- P‑glycoprotein (P‑gp) involvement: Ibrutinib is a substrate for intestinal P‑gp efflux pumps. Co‑administration with strong P‑gp inhibitors (e.g., verapamil) can raise exposure by up to 30%.
Distribution: Where the Drug Travels
After absorption, Ibrutinib displays a high volume of distribution (~ 250 L), reflecting extensive tissue binding. Protein binding is >97%, mainly to albumin and α‑1‑acid glycoprotein.
Key distribution points:
- Peripheral blood mononuclear cells (PBMCs): Ibrutinib concentrates in the very cells where BTK resides.
- Central nervous system: Limited penetration due to P‑gp activity at the blood‑brain barrier; not a primary concern for CLL but relevant when treating CNS‑involved lymphoma.
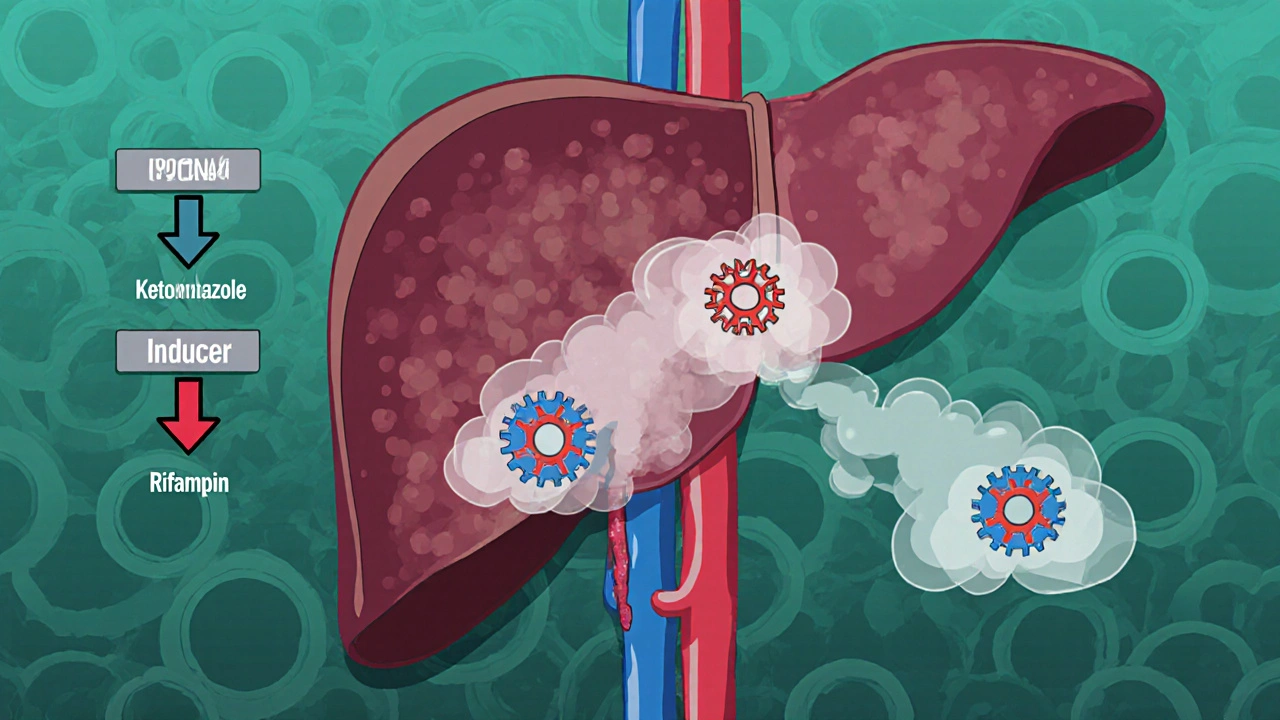
Metabolism: The CYP3A4 Connection
The liver is the principal site of metabolism. Ibrutinib is extensively metabolized by the cytochrome P450 isozyme CYP3A4, generating several inactive metabolites, the most abundant being PCI‑45227. Minor pathways involve CYP2D6 and CYP2C9, but they contribute less than 10% of total clearance.
Clinical implications:
- Strong CYP3A4 inhibitors (ketoconazole, itraconazole, clarithromycin) can increase Ibrutinib AUC by 2‑ to 3‑fold, raising the risk of grade ≥ 3 adverse events such as atrial fibrillation and bleeding.
- Strong CYP3A4 inducers (rifampin, carbamazepine, St. John’s wort) can cut exposure by ~50%, potentially compromising disease control.
- Therapeutic drug monitoring (TDM) is not routine but may be considered in patients on interacting medications.
Elimination: How the Body Clears Ibrutinib
The mean apparent clearance (CL/F) is about 30‑40 L/h. Approximately 80% of the dose is eliminated via feces (mainly as metabolites), while the remaining 20% is excreted in urine, again largely as metabolites.
The most critical factor when prescribing Ibrutinib is understanding its half‑life. The terminal elimination half‑life ranges from 4 to 6 hours, but due to irreversible BTK binding, the pharmacodynamic effect persists for 24 hours, justifying once‑daily dosing.
Factors That Modify Ibrutinib Pharmacokinetics
Beyond drug‑drug interactions, several patient‑specific variables influence PK:
- Age: No dose adjustment needed up to 85 years; elderly patients may experience higher plasma concentrations due to decreased hepatic blood flow.
- Sex: Minor differences (<10%) observed, not clinically relevant.
- Renal impairment: Mild to moderate kidney disease does not require a dose change; severe impairment (CrCl <30 mL/min) has limited data, so cautious use is advised.
- Hepatic impairment: Moderate (Child‑Pugh B) raises AUC by ~ 30%; recommended to reduce dose to 140 mg daily. Severe impairment (Child‑Pugh C) is contraindicated.

Clinical Dosing Recommendations Based on PK
The standard adult dose for CLL and MCL is 420 mg once daily (or 560 mg for certain protocols). Dose reductions to 280 mg or 140 mg are advised when:
- Co‑administration with a strong CYP3A4 inhibitor is unavoidable; reduce to 140 mg.
- Patients develop grade ≥ 3 toxicities (e.g., severe diarrhea, neutropenia, bleeding).
- Moderate hepatic impairment is present.
After any dose change, steady‑state is typically reached within 3-4 days, allowing clinicians to assess response and toxicity promptly.
Comparison with Other BTK Inhibitors
| Drug | Bioavailability | Half‑life | Primary Metabolism | Typical Dose |
|---|---|---|---|---|
| Ibrutinib | ~97% | 4-6 h (pharmacokinetic) | CYP3A4 | 420 mg QD |
| Acalabrutinib | ~81% | 1 h | CYP3A4 | 100 mg BID |
| Zanubrutinib | ~94% | 2-4 h | CYP3A4 | 160 mg BID |
While all three agents share CYP3A4 metabolism, Ibrutinib’s longer half‑life and higher bioavailability allow once‑daily dosing, which can improve adherence. However, the same metabolic pathway makes it more vulnerable to drug interactions compared with the newer, shorter‑acting inhibitors.
Managing Common Adverse Effects Through PK Insight
Because exposure correlates with toxicity, clinicians can mitigate side‑effects by adjusting dose or avoiding interacting drugs:
- Bleeding risk: Anticoagulants (warfarin, DOACs) add to platelet dysfunction. If combined therapy is essential, reduce Ibrutinib to 140 mg and monitor INR or drug‑specific assays.
- Atrial fibrillation: Higher plasma levels double the incidence. Use dose reduction or switch to a BTK inhibitor with a shorter half‑life if arrhythmia persists.
- Diarrhea and rash: Usually dose‑dependent; symptomatic treatment plus a temporary dose hold of 3‑7 days often suffices.
Future Directions: PK‑Guided Personalization
Research is exploring genotype‑guided dosing. Polymorphisms in CYP3A4*22 and ABCB1 (P‑gp) can shift exposure by up to 20-30%. While routine testing is not yet standard, high‑risk patients (e.g., on multiple interacting meds) may benefit from early PK assessment.
Does food affect Ibrutinib absorption?
Ibrutinib has ~97% oral bioavailability, and a high‑fat meal only raises exposure by about 10%, which is not clinically significant. Patients can take it with or without food.
What are the major drug interactions to watch for?
Strong CYP3A4 inhibitors (ketoconazole, clarithromycin) markedly increase Ibrutinib levels and require dose reduction to 140 mg. Strong CYP3A4 inducers (rifampin, carbamazepine) lower exposure and may necessitate dose escalation or switching agents.
How is Ibrutinib cleared from the body?
Approximately 80% of the dose is eliminated via feces as metabolites, with the remaining 20% excreted in urine. The mean apparent clearance is 30‑40 L/h.
Do I need to adjust the dose for elderly patients?
No routine dose reduction is required solely for age. However, monitor for increased toxicity, especially if liver function is impaired.
Can I take Ibrutinib with anticoagulants?
Concurrent anticoagulation raises bleeding risk. If unavoidable, reduce Ibrutinib to 140 mg, monitor for signs of bleeding, and consider using a BTK inhibitor with a shorter half‑life.
Ok, so the whole Ibrutinib hype is kinda overblown. The bioavailability numbers look slick, but most docs forget that real‑world patients are on a cocktail of meds that can flip the PK curve upside‑down.
And let’s be real, who’s got the time to monitor every CYP3A4 interaction? It’s a nightmare in a community clinic.
Bottom line: the dosing guidelines are fine on paper, but in practice they’re a moving target.
When prescribing Ibrutinib you need to treat the PK profile like a checklist, not a decorative footnote. First, the near‑perfect oral bioavailability means you can count on the dose getting into the bloodstream, but that only matters if the patient isn’t chewing the capsule or vomiting. Second, the 1‑2 hour Tmax tells you the drug peaks quickly, so any missed dose will show up as a noticeable dip in exposure within a few hours. Third, remember that CYP3A4 is the main metabolic gatekeeper; a strong inhibitor like ketoconazole can double or triple the AUC, which directly translates into higher rates of atrial fibrillation, bleeding, and severe diarrhea. Fourth, the flip side is that inducers such as rifampin slice the exposure in half, potentially rendering the therapy ineffective against CLL. Fifth, the irreversible BTK binding gives you a pharmacodynamic half‑life that lasts far beyond the plasma half‑life, which is why once‑daily dosing works despite a 4‑6 hour terminal half‑life. Sixth, food only nudges the AUC by about ten percent, so you don’t have to enforce fasting, but advise patients to avoid very high‑fat meals if they notice GI upset. Seventh, the drug’s volume of distribution is huge – roughly 250 L – indicating deep tissue penetration, but the high protein binding (>97 %) means that changes in albumin levels can shift the free fraction. Eighth, hepatic impairment is the real red flag; severe cases demand dose reduction because the liver can’t clear the metabolites efficiently. Ninth, renal function matters less for Ibrutinib, but you still have to watch for metabolite accumulation in chronic kidney disease. Tenth, co‑administration with P‑gp inhibitors like verapamil can raise exposure by up to thirty percent, so you may need to tweak the dose or switch the interacting drug. Eleventh, therapeutic drug monitoring isn’t routine, but in complex polypharmacy scenarios it becomes a valuable safety net. Twelfth, always counsel patients about the bleeding risk, especially if they’re on anticoagulants or antiplatelet agents. Thirteenth, keep an eye on cardiac monitoring because atrial fibrillation can appear abruptly after a drug‑drug interaction spikes the levels. Fourteenth, educate the care team that the adverse‑event profile is dose‑dependent, so dose reductions are a legitimate tool, not a failure. Fifteenth, document every interaction and dose adjustment in the EMR to protect both the patient and the prescriber from liability. In short, Ibrutinib is a powerhouse when used correctly, but it demands a disciplined approach to managing interactions, organ function, and adverse‑event surveillance.
Everyone acts like Ibrutinib is just another pill, but have you ever wondered why the pharma giants keep pushing a drug that hinges so heavily on CYP3A4? It's as if they're counting on us to stay blind to the hidden network of enzyme modulators that silently reshape our biology. The truth is buried under layers of clinical trial fluff, where selective data is cherry‑picked to paint a picture of safety while the real risk matrix remains concealed. If you think the irreversible BTK binding is a blessing, consider that it also locks the drug into a chemical partnership we can’t reverse, giving the manufacturers a silent grip on patient outcomes. The metabolism pathways whisper a secret: a few concoctions of over‑the‑counter supplements can sabotage the efficacy, and the big pharma narrative never mentions that. In a world where every molecule is a potential lever for control, Ibrutinib is just another cog that shows us how deep the rabbit hole goes.
We must ask: who decides the dosing thresholds, and why are they so flexible when it comes to hepatic impairment? The answer may be hidden in the boardroom, not in the peer‑reviewed article.
This drug hits the BTK target hard and leaves side‑effects in its wake.
Honestly!!! The pharmacokinetic data on Ibrutinib is presented as if it were carved in stone; yet the reality is far messier, more chaotic, more human!!! One must ask: does the 97% bioavailability truly reflect real‑world adherence, or is it a glossy veneer crafted by the industry?? The interplay of CYP3A4 inhibition is not just a biochemical footnote; it is a profound reminder that our bodies are ecosystems of interaction-ever‑shifting, ever‑responsive!!! If we ignore the subtle nuances of protein binding >97%, we betray the very principle of precision medicine; we become mere pawns in a larger pharmacoeconomic game!!! Therefore, dosing adjustments should not be an afterthought but a deliberate, philosophically grounded practice!!!
While Corrine raises valid points about the complexities of Ibrutinib metabolism, the clinical trials do provide robust data on safety thresholds. The reported 97% bioavailability, for instance, comes from controlled studies with strict adherence protocols, which may not fully translate to outpatient settings, but they still offer a useful benchmark. Moreover, the impact of CYP3A4 inhibitors has been quantitated, allowing clinicians to make evidence‑based dose reductions when necessary. It's also worth noting that severe hepatic impairment is the primary scenario where dose adjustment is mandatory, aligning with pharmacokinetic predictions. In short, the guidelines aren't arbitrary-they're grounded in measurable pharmacodynamic outcomes, even if real‑world variability demands clinician vigilance.
From an Indian perspective, we often see patients juggling Ayurvedic herbs alongside mainstream chemo-something the Western guidelines barely touch. Strong CYP3A4 inducers like St. John’s wort are common in our community, and they can slash Ibrutinib levels dramatically; doctors must ask patients about herbal supplements before scribbling a prescription. Also, the diet here can be high in fat, which modestly raises AUC, so counseling on meal timing becomes essential. The pharmacokinetic principles are universal, but the cultural context changes how we apply them, and that nuance is missing from many official recommendations.
Great rundown! 🙌 Loved the clear break‑down of absorption and metabolism. The food‑effect note is super helpful-now I’ll tell my cousin to take it with a light snack 😅. Also, the tip about checking for CYP3A4 inhibitors saved me from a potential bleed, thank you! 👍